 医薬品分子は、メチル基や塩素原子を導入したり、複素環のヘテロ原子の位置を変えたりする微小な構造変化により、活性が10倍以上向上するような事例が知られている。ただ、分子構造だけからこうした活性の変化を予測することは困難であり、結合自由エネルギー計算によるアシストが有効だとされる。FEP法とTI法は、一つの受容体に作用する複数のリガンドの結合自由エネルギーの差を精密に求めることが可能。リガンド構造を調整した際の活性の変化を実験せずに評価することができる。
医薬品分子は、メチル基や塩素原子を導入したり、複素環のヘテロ原子の位置を変えたりする微小な構造変化により、活性が10倍以上向上するような事例が知られている。ただ、分子構造だけからこうした活性の変化を予測することは困難であり、結合自由エネルギー計算によるアシストが有効だとされる。FEP法とTI法は、一つの受容体に作用する複数のリガンドの結合自由エネルギーの差を精密に求めることが可能。リガンド構造を調整した際の活性の変化を実験せずに評価することができる。
モルシスが「MOE」で結合自由エネルギー計算
リガンド活性の変化を精密に評価、GPU対応のAMBER利用
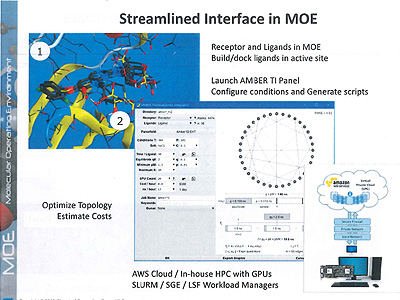
2019.08.22−モルシスが提供している統合計算化学システム「MOE」(米CCG社製)最新バージョンに搭載された新機能が注目されている。医薬品分子設計において、受容体とリガンドとの結合自由エネルギーを高精度に計算する技術で、分子構造の微小な変化による活性の向上を予測することができるため、リードオプティマイゼーションを行う際に大きな武器となる。米カリフォルニア大学などで開発されている分子動力学法(MD)プログラム「AMBER」の最新版で利用できるサーモダイナミックインテグレーション法(TI)を利用したもの。この分野は、米シュレーディンガーがフリーエナジーパータベーション法(FEP)に基づく「FEP+」でシェアを握っており、その勢力争いも注目される。
医薬品分子は、メチル基や塩素原子を導入したり、複素環のヘテロ原子の位置を変えたりする微小な構造変化により、活性が10倍以上向上するような事例が知られている。ただ、分子構造だけからこうした活性の変化を予測することは困難であり、結合自由エネルギー計算によるアシストが有効だとされる。FEP法とTI法は、一つの受容体に作用する複数のリガンドの結合自由エネルギーの差を精密に求めることが可能。リガンド構造を調整した際の活性の変化を実験せずに評価することができる。
ベースになるのはMD計算だが、計算量が大きいことが問題で、長く実用化が図られてこなかったという。ところが、シュレーディンガーがGPU(グラフィックプロセッサー)をフル活用した「FEP+」を2015年に製品化したことで関心に火がつき、AMBERも最新版の「AMBER 18」でGPU対応のTI法を正式にサポートした。
今回の「MOE 2019.01」に搭載されたのは、MOEのGUI(グラフィカルユーザーインターフェース)を利用してAMBERによるTI計算を実行する機能。MOE上で受容体とリガンドの構造を作成・準備し、TI計算のためのジョブスクリプトを生成、オンプレミスあるいはクラウド環境でGPU対応計算を実行し、その結果をMOEに戻して解析を行う。
TI計算はエラー率が高く、実行が難しいが、GPUやノード数などの利用可能な計算リソースに基づき、予想されるエラーを最小化する最適なシミュレーション計画を立てることで対応。また、静電気力とファンデルワールス力の不均衡によって計算が不安定になることがあるため、AMBERのソフトコアポテンシャルの最適化を図っているという。
クラウドサービスとして、アマゾンウェブサービス(AWS)に対応しており、GPUマシンなどを社内に用意する手間をかけることなく、すぐに計算することが可能。費用も手ごろで、1件当たりの計算は100ドルほどになるという。
そのほか、MOE 2019.01の新機能としては、指定したpH範囲での立体配座依存性などの新しいタンパク質物性推算機能、SAR(構造活性相関)解析のためのウェブアプリケーションMOEsaicの機能、NMRデータによる立体配座分布解析(溶液中でのリガンド配座のアンサンブル決定)などの機能強化が図られている。
******
<関連リンク>:
モルシス(トップページ)
https://www.molsis.co.jp/
モルシス(MOE 製品情報ページ)
https://www.molsis.co.jp/lifescience/moe/